One of my primary motivations for setting up this blog was to have a centralized place for sharing how I am using the combination of the RDKit and the IPython notebook in teaching and research. There are many great resources for learning how to use the RDKit python bindings, including the indispensible “Getting Started width the RDKit in Python”, Greg Landrum’s RDkit blog, and a particularly nice example from the ChEMBL-og.
Our first step is to import the module that we will be using:
from rdkit import Chem
Special imports for drawing molecules:
from rdkit.Chem.Draw import IPythonConsole #Needed to show molecules
from rdkit.Chem.Draw.MolDrawing import MolDrawing, DrawingOptions #Only needed if modifying defaults
Modify default molecule drawing settings:
DrawingOptions.bondLineWidth=1.8
Create a molecule object from a SMILES string:
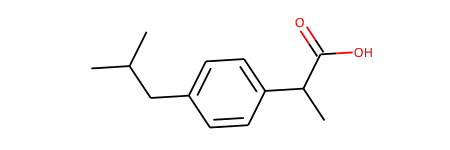
ibu = Chem.MolFromSmiles('CC(C)Cc1ccc(cc1)C(C)C(=O)O')
Display the molecule in the notebook:
ibu

The molecule object can now be queried to get various properties
ibu.GetNumAtoms()
Anything seem wrong here? Let's print out a text representation of the molecule and take a closer look:
print Chem.MolToMolBlock(ibu)
We see that the hydrogen atoms are implicit in this representation of the molecule. They were not included in the SMILES string and are still not present in the connection table.
DrawingOptions.includeAtomNumbers=True
ibu

If we are interested in doing anything that requires atomic coordinates, we probably want an all-atom representation of the molecule. We can make the hydrogen atoms explicit by adding them (and creating a new molecule in the process). We need more functions than provided in the base Chem module, so we need to import the expanded functionality of AllChem.
from rdkit.Chem import AllChem
ibuH = AllChem.AddHs(ibu)
DrawingOptions.includeAtomNumbers=False
ibuH

print Chem.MolToMolBlock(ibuH)
ibuH.GetNumAtoms()
Our all-atom represntation is now suitable for many purposes, such as calculating properties that depend only upon atom-types and connectivity. One of these is the total polar surface area (TPSA):
Chem.rdMolDescriptors.CalcTPSA(ibuH)
For other properties, we will need more information about our molecule.
We can save the molecule for later use in a python format called a pickle:
import cPickle as pickle
pickle.dump(ibuH, open('ibuH.pkl','wb'))